
EU AI Act in healthcare: Beyond compliance to strategic resilience

The EU AI Act does something unprecedented in healthcare: it elevates high-risk AI to the regulatory expectations of clinical-grade systems, rather than treating them as standard software.
This shift has immediate consequences. Systems that influence diagnosis, triage, or treatment are no longer evaluated only on performance, but on traceability, explainability, and accountability.
For many organisations, this is where the real challenge begins: not with the regulation itself, but with the systems they’ve already built. The clock is already ticking toward the 2026 and 2027 compliance deadlines, and retrofitting an AI system is always harder than building it right the first time.
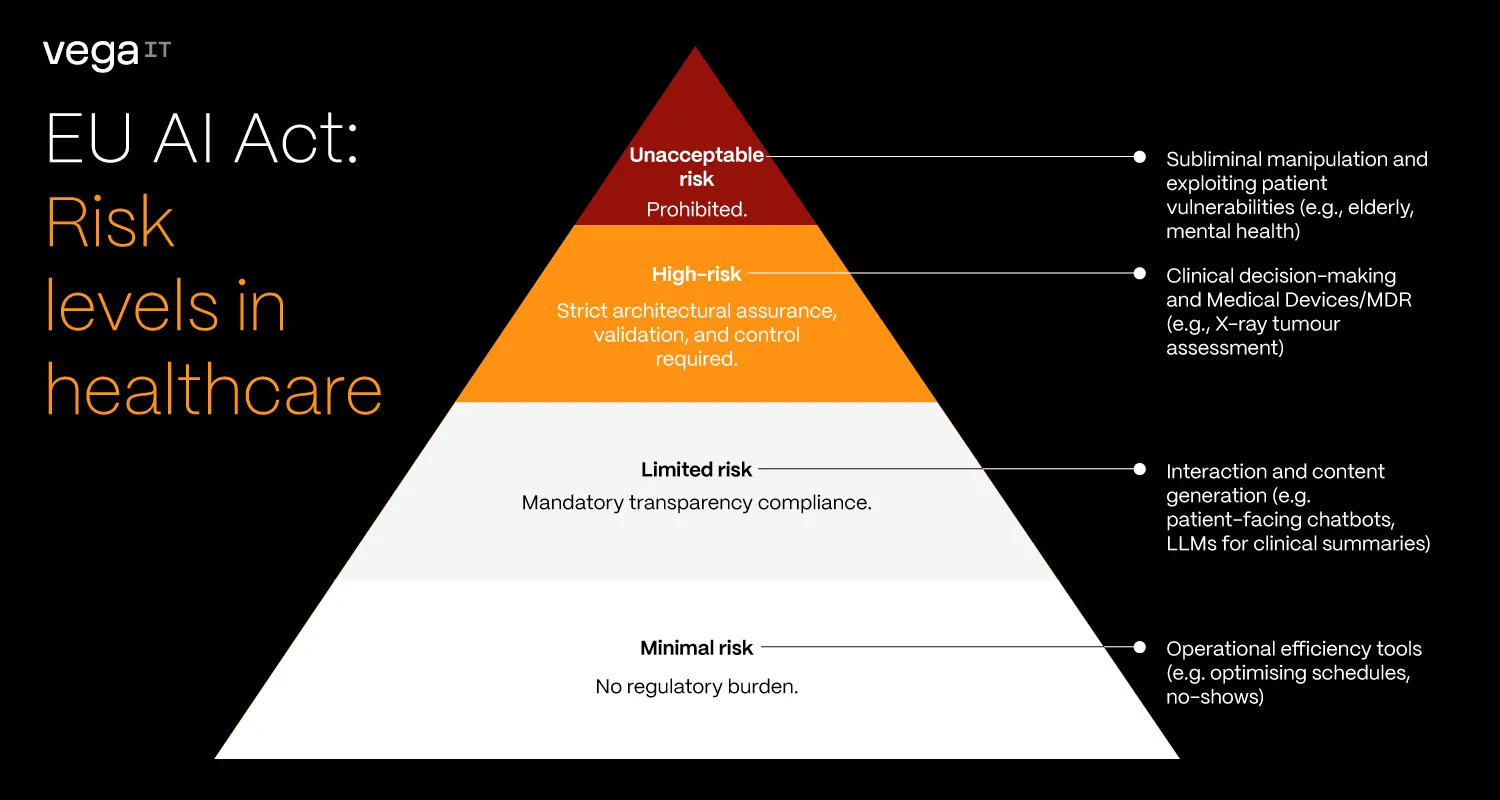
The risk hierarchy: Is your AI a "calculator", “communicator” or a "co-pilot"?
The AI Act uses a risk-based approach. In practice, systems that influence clinical decisions are treated fundamentally differently from those that don't.
- Minimal risk systems (the calculators): Tools for operational efficiency, like predicting patient no-shows or optimising schedules. If they fail, the impact is financial. The AI Act imposes virtually no regulatory burden here, leaving you free to innovate rapidly.
- Limited risk systems (the communicators): Patient-facing chatbots or LLMs drafting clinical summaries. The core mandate is transparency. Users must explicitly know they are interacting with, or reading content generated by, an AI.
- High-risk systems (the co-pilots): Systems operating inside clinical decision-making, like assessing an X-ray for a tumour. Crucially, if your software is already a Medical Device (MDR), it automatically defaults to this category. These require strict architectural assurance, validation, and control.
- Unacceptable risk (the red lines): These are outright banned. In a healthcare context, this includes AI systems that deploy subliminal techniques to alter behaviour, or exploit the vulnerabilities of specific patient groups (e.g., mental health patients or the elderly) in ways that cause harm. While legitimate MedTech rarely crosses this line, documenting that your system does not fall into this category is step zero of compliance.
The roadmap: How much time do you have?
The AI Act is a marathon, not a sprint.
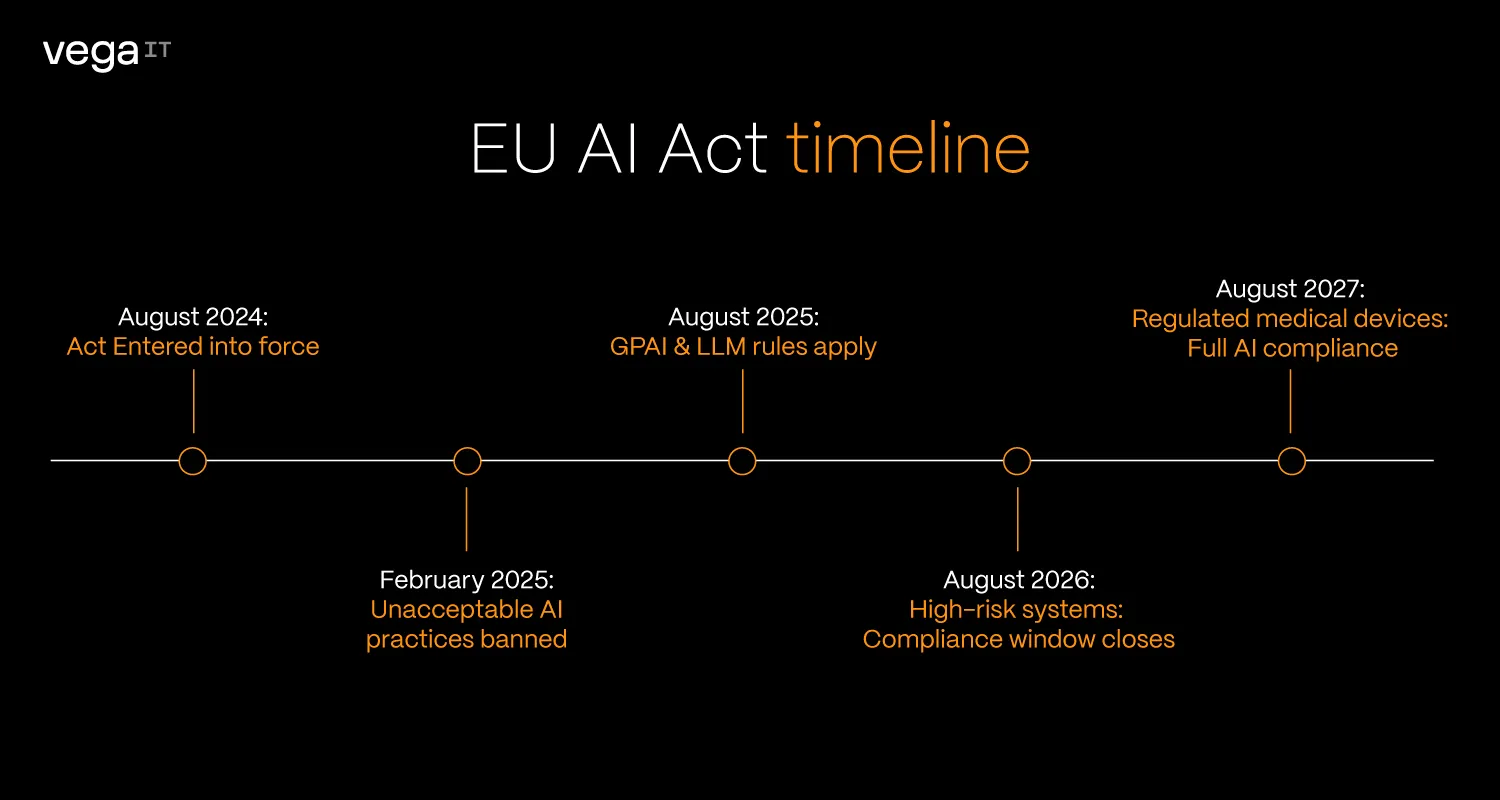
EU AI Act timeline:
- August 2024:The Act entered into force.
- February 2025: Prohibitions on unacceptable AI practices became officially applicable.
- August 2025: Rules for General-Purpose AI (GPAI), including large language models, took effect.
- August 2026: Most high-risk systems are about to fall under full compliance obligations.
- August 2027: The "Big One." High-risk AI that is already part of regulated medical devices must be fully compliant.
Most organisations focus on these deadlines. The real risk is that by the time they arrive, underlying systems will not be ready to support them.
Non-compliance is not marginal. Under Article 99, fines can reach up to €35 million or 7% of global annual turnover, whichever is higher. In healthcare, this is not just a financial penalty, it is a signal that patient safety is a regulatory boundary, not a negotiable outcome.
The end of “black box” medicine
In traditional AI systems, outputs were often not explainable in practical terms: data goes in, prediction comes out, and the reasoning remains opaque.
The AI Act changes that expectation for clinical outputs.
Practical example:
Imagine a Radiology AI that flags a chest X-ray for pneumonia.
- Without the AI Act: The software simply states "90% chance of Pneumonia." The doctor has to take its word for it.
- With the AI Act: The software often needs to provide features like "Saliency Map" (a heatmap) showing exactly which pixels in the lung area triggered the alert. This allows the doctor to verify the AI's logic in seconds, turning a "mysterious oracle" into a transparent assistant.
Transparency goes beyond the UI
However, clinical explainability is only one piece of the puzzle. The AI Act demands transparency on multiple layers:
- Business: Leaders need to know exactly where AI is deployed, ensure continuous system monitoring, and verify third-party vendor compliance.
- Engineering: Teams must have full visibility into models, prompts, and data sources.
- End-users: Patients and clinicians must explicitly know if and when AI is being used.
Compliance is an architectural decision
Because transparency is required across all these layers, teams that succeed with the AI Act treat compliance as a design principle, not a validation step.
For high-risk AI, the law demands continuous proof that the system is traceable, reliable, rigorously tested, monitored, and reviewable by humans. Translating those legal requirements into engineering practice means:
In practice, that means:
- Built-in traceability – every decision can be reconstructed and audited
- Modular AI components – models can be updated without disrupting clinical workflows
- Clear data lineage – training and inference data are controlled and verifiable
Without standardised data (FHIR, openEHR), auditability and model governance become difficult to maintain at scale.
Retrofitting these capabilities later isn't just complex, it is often financially prohibitive compared to building them in from the start.
This is where the real divide will emerge: between organisations that build AI features, and those that build clinical-grade systems.
The regulatory trifecta: The gearbox of EU healthcare
The AI Act does not operate in isolation. It works alongside:
1. EHDS (European Health Data Space): enabling structured, cross-border data access
2. Product Liability Directive (PLD): formally treating software as a product with clear accountability
Together, they redefine the environment:
Data becomes more accessible, but also more governed
Innovation becomes easier, but liability becomes clearer
Success in this landscape depends on aligning all three, not treating them as separate compliance tracks.
What healthcare leaders should focus on today
Reclassify your AI portfolio
Take a thorough inventory of every algorithm in your ecosystem. The AI Act classification hinges on intent. You need to distinguish between "administrative calculators" (low risk) and those acting as Medical Devices (high risk). Identifying these now prevents a massive regulatory "recalculating" fee in 2027.
Standardise your data
If the AI Act is the law, then high-quality data is the evidence. Start using global standards like FHIR or openEHR today. Without these frameworks, clinical data suffers from inconsistent wording and fragmentation, making it significantly harder to train reliable AI, run accurate analytics, or conduct compliance audits.
Standardisation is the difference between a messy pile of notes and a perfectly organised digital archive that is "audit-ready" for the strict data governance requirements of the Act.
Design for human oversight
Compliance isn't just a back-end issue; it’s a UI/UX challenge. You must ensure your interfaces allow clinicians to easily understand the AI’s logic and, crucially, override its suggestions. Under the new law, "human-in-the-loop" is no longer a design choice, it’s a legal mandate.
Leverage regulatory sandboxes
Regulatory sandboxes and research exemptions offer a controlled, risk-free environment to test your innovations under regulatory guidance. However, waiting to engage with these mechanisms until the 2027 enforcement deadline approaches removes the strategic advantage of refining your architecture early.
Trust as the ultimate clinical outcome
The EU AI Act is often framed as a constraint on innovation. In reality, it is more likely to act as a filter. It will separate systems that produce outputs from systems that can be trusted in clinical environments.
Compliance is not just about avoiding penalties. It is about proving that AI can operate safely where it matters most – a standard that requires responsible healthcare software development.
The organisations that adapt early won’t just be compliant, they will define what “clinical-grade AI” means in practice.
