
Is your health app a medical device?
Understanding SaMD, classification, and compliance

The journey of developing digital products in the health, beauty, and wellness space often leads to a pivotal question: is this app a medical device? The answer can redefine an entire project’s trajectory. It shapes regulatory obligations, development strategies, and even go-to-market plans.
Recognising whether your solution falls into the category of medical device software or remains a general wellness tool is not just semantics; it’s the first step in navigating the complex world of digital health regulation.
In this article, we’ll unpack the implications of medical device classification for software creators. We'll look into the benefits as well as the additional burdens that come with a medical device designation. Beyond the what and the how, we'll also examine the fundamental "why" – explaining why these regulations are necessary for ensuring patient safety and product efficacy.
What is a medical device?
This is not a simple question, and it rarely comes with a thrilling answer. The official definition is a legal one, set by regulatory bodies, which means the exact wording can differ depending on the region.
At its core, though, the meaning stays the same: a medical device is any product, including software, intended for medical use in humans that does not achieve its primary purpose through pharmacological, immunological, or metabolic means.
What is software as a medical device (SaMD)?
Medical device software refers to software that qualifies as a medical device in its own right, intended for one or more medical purposes without being part of a physical hardware medical device. These apps can operate on a wide range of general-purpose platforms, including smartphones and tablets, or in the cloud.
As with medical devices in general, definitions can vary depending on the regulatory body (e.g. the FDA in the US or the MDR in the EU). Still, when it comes to software as a medical device, two primary factors usually determine its classification:
- Intended use
- Medical purpose
What do we mean by “intended use”?
The term “intended use” refers to the medical purpose for which the manufacturer or developer explicitly claims the software will be used. If the product's marketing, labelling, or instructions state that it is for the diagnosis, treatment, mitigation, or prevention of a disease, it falls under medical device classification and is subject to digital health regulation.
It’s important to note that the way users apply software on their own does not automatically make it a medical device. For example, if someone uses a wellness app in a creative or unintended way for medical purposes, that alone doesn’t change its status. However, if the manufacturer actively promotes or encourages such use, the software could then be reclassified as a medical device.
When does software have a medical purpose?
This part is a little more complex because it requires a detailed look at both the software’s functionality and its intended use. If the software provides information that is meant to support a medical decision, it is likely to be considered medical device software. On the other hand, simply displaying raw information or markers is not, in itself, a medical purpose. But once the software interprets that data, offers insights, or suggests next steps in diagnosis or treatment, it crosses into the domain of software as a medical device (SaMD).
A commonly used example highlights the difference. A fitness tracker that displays heart rate data is usually not classified as a medical device. However, if the same tracker analyses heart rate data and issues an alert about a possible cardiac arrhythmia, it would fall under software as a medical device and be subject to digital health regulation.
Of course, real-world scenarios are rarely this clear-cut. Many cases fall into a grey area, which is why the guidelines from individual regulatory bodies need to be consulted to provide a final medical device classification.
Still, the core questions are always the same:
- What does the software do?
- What is the intended medical purpose of the software?
- What medical claims are made by the manufacturer?
- Does the software provide information intended to influence medical decisions?
- What is the level of risk to the patient if the software fails or provides incorrect output?

Intended use in action: The Mettle App
Our work on Mettle (co-founded by Bear Grylls) is a perfect example of a general wellness tool. Because it focuses on everyday mental fitness and stress management rather than diagnosing or treating clinical conditions, it falls completely outside the scope of SaMD regulations.
Learn moreMedical device classification
So, you’ve determined that your software is indeed a medical device. What comes next? The real work begins with medical device classification, which is perhaps the single most important step in the regulatory process. While the manufacturer initiates the classification process, it is ultimately the responsibility of the relevant regulatory body or a designated third party to validate and approve that classification.
The classification of any software as a medical device (SaMD) is determined by its potential risk to the patient. This risk is assessed through two primary factors:
- The significance of the information provided by the software
- The state of the healthcare situation or condition it is addressing
Based on these dimensions, SaMD products can generally be grouped into the following categories:
- Low Risk (Class I): SaMD intended to inform or guide clinical management for a non-serious condition.
- Medium Risk (Class II): SaMD intended to treat or diagnose a non-serious condition, or to inform or drive clinical management for a serious condition.
- High Risk (Class III): SaMD intended to treat or diagnose a critical condition. These devices are subject to the most rigorous scrutiny and require extensive clinical evidence to demonstrate safety and efficacy.
It’s important to note that classification systems vary by region. For example, under the EU’s Medical Device Regulation (MDR), a four-class system is used, with additional subcategories for Class I devices. In the EU, independent third-party organisations known as Notified Bodies handle reviews and approvals, while in the US, this role falls to the FDA. The EU relies on a rule-based system for classification, whereas the FDA focuses more on intended use and benchmarking risk against products already on the market.
This is why understanding your target market is critical at this stage – different regulators can classify the same software in different ways.
Classifying software as a medical device isn’t just regulation – it’s a strategic investment in your product’s future. Choosing your technical partner wisely can transform this challenging process into a smoother, more manageable journey.
What comes next?
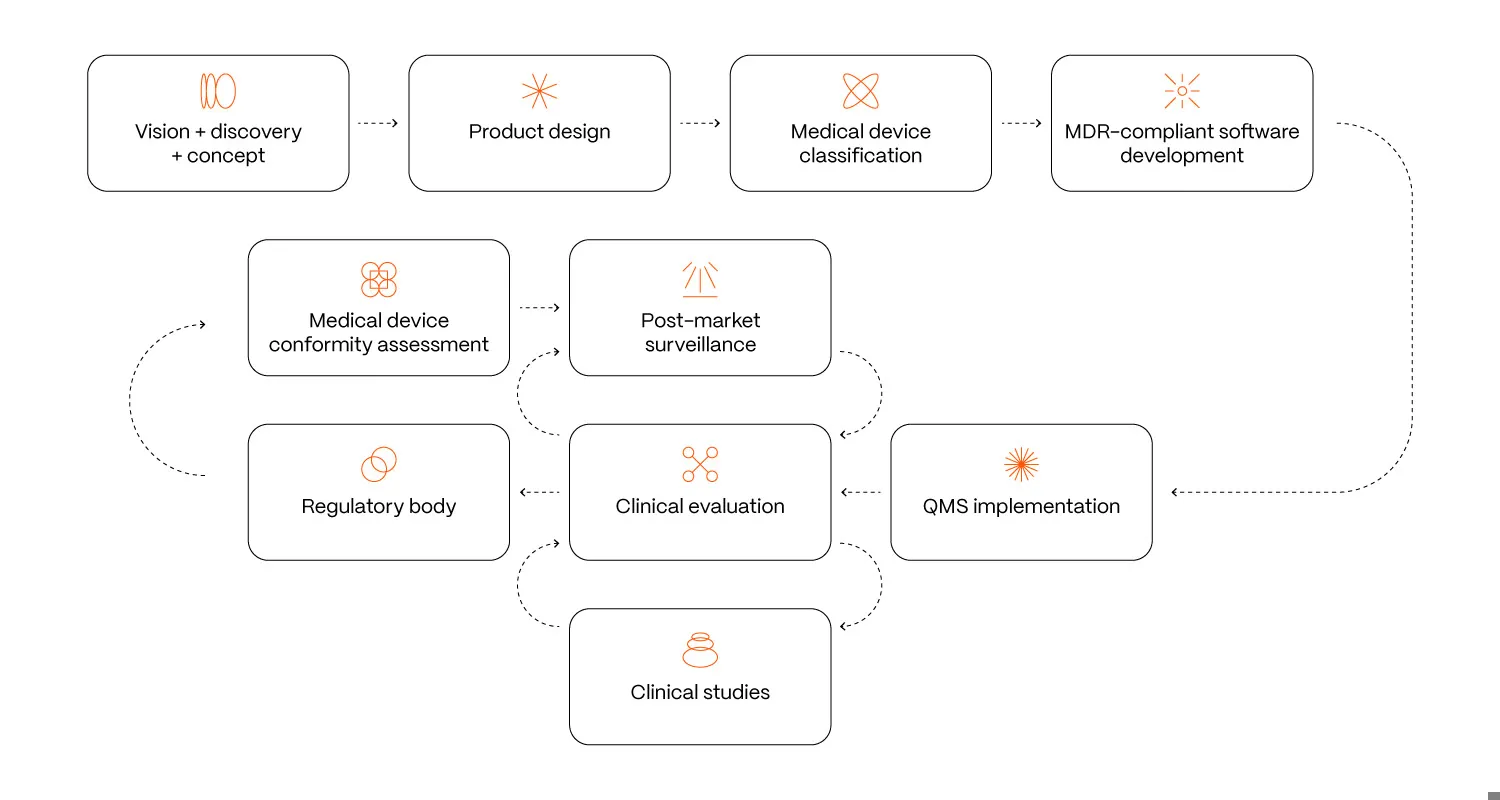
Reaching a medical device classification is not the finish line but the beginning of a demanding journey that transforms a simple software project into a fully regulated medical product. This designation introduces significant additional obligations and costs, which need to be understood from the outset.
Once your software is designated as Software as a Medical Device (SaMD), you are legally required to implement a robust quality management system to control every step of the development, manufacturing, and post-market life.
This involves:
- Creating extensive technical documentation, covering everything from design inputs to risk management.
- Conducting rigorous testing, including thorough software validation and, for higher-risk devices, clinical validation to prove safety and efficacy.
- Preparing and submitting detailed regulatory documentation to the relevant authorities for market clearance.
- Carrying out continuous post-market surveillance to track real-world performance and report any adverse events.
These ongoing compliance requirements add significant cost, time, and operational complexity, often reshaping the way a software company functions once it steps into the regulated medical space.
Why SaMD classification matters
The process of classifying and regulating software as a medical device is demanding, but it’s far more than a regulatory burden. It’s a strategic investment in your product’s future. The benefits of correct SaMD classification are significant and directly influence your ability to succeed in the competitive digital health market.
The most important benefit is market access. Without the proper classification, a product cannot be legally sold for a medical purpose in major regions such as the US and the EU. Classification also acts as a powerful signal to healthcare providers, hospitals, and insurers that your medical device software has been rigorously assessed for safety, efficacy, and quality. This is often a prerequisite for a hospital or healthcare system to even consider integrating your software, proving that your digital product and solution development for healthcare meets the highest standards for market entry and trust.
In addition to building trust with both patients and healthcare professionals, there are also practical business reasons patients would prefer to use a certified medical device. In many healthcare systems, a SaMD rating is essential for a product to be eligible for reimbursement by insurance companies, which is critical for widespread adoption.
Finally, compliance reduces risk. Having a product reviewed and approved as safe and effective not only protects patients, it also provides a safeguard against liability claims in cases of potential harm.
